Effect of apolipoprotein E and Alzheimer’s disease related pathologies
The apolipoprotein E gene (APOE) is the strongest genetic risk factor for AD. One copy of the ε4 isoform of APOE increases AD risk by ~3.7 fold whereas 2 copies of the ε4 isoform increases risk by ~12-fold relative to the ε3 allele. One copy of the ε2 allele of APOE decreases AD risk by ~0.6. The ε4 allele also increases risk for CAA. While there are several ways that apoE may be contributing to AD risk, our data along with data from other labs demonstrates that a major mechanism by which apoE contributes to AD risk is via apoE’s effects on both soluble Aβ clearance as well as directly affecting Aβ aggregation. Over the last 7 years, we have also found that apoE strongly influences tau-mediated neurodegeneration via a mechanism in part linked to effects on the brain’s innate and adaptive immune system. This appears to be an important mechanism by which apoE can influence not only risk but also progression of AD and primary tauopathies. These findings provide novel insights into the links between apoE and neurodegenerative diseases. All the work referenced below was carried out in my lab and all experiments, personnel, data analysis, and writing of manuscripts were under my direction.
Publications

Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy
March 8, 2023
Chen X, Firulyova M, Manis M, Herz J, Smirnov I, Aladyeva E, Wang C, Bao X, Finn MB, Hu H, Shchukina I, Kim MW, Yuede CM, Kipnis J, Artyomov MN, Ulrich JD, Holtzman DM. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. 2023 Mar 8. doi: 10.1038/s41586-023-05788-0. Epub ahead of print. PMID: 36890231.

Overexpressing low-density lipoprotein receptor reduces tau-associated neurodegeneration in relation to apoE-linked mechanisms
August 4, 2021
Shi Y, Andhey PS, Ising C, Wang K, Snipes LL, Boyer K, Lawson S, Yamada K, Qin W, Manis M, Serrano JR, Benitez BA, Schmidt RE, Artyomov M, Ulrich JD, Holtzman DM. Overexpressing low-density lipoprotein receptor reduces tau-associated neurodegeneration in relation to apoE-linked mechanisms. Neuron. 2021 Aug 4;109(15):2413-2426.e7. doi: 10.1016/j.neuron.2021.05.034. Epub 2021 Jun 21. PMID: 34157306; PMCID: PMC8349883.

Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia
May 19, 2021
Wang C, Xiong M, Gratuze M, Bao X, Shi Y, Andhey PS, Manis M, Schroeder C, Yin Z, Madore C, Butovsky O, Artyomov M, Ulrich JD, Holtzman DM. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron. 2021 May 19;109(10):1657-1674.e7. doi: 10.1016/j.neuron.2021.03.024. Epub 2021 Apr 7. PMID: 33831349; PMCID: PMC8141024.
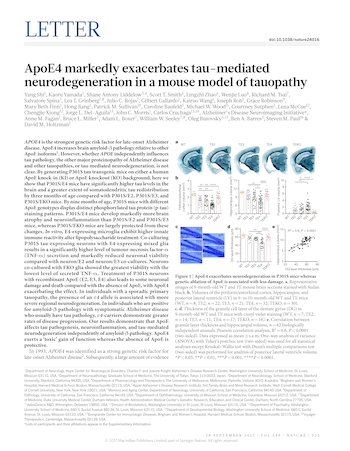
ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy
September 28, 2017
Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, McCue L, Xiong C, Del-Aguila JL, Morris JC, Cruchaga C; Alzheimer’s Disease Neuroimaging Initiative; Fagan AM, Miller BL, Boxer AL, Seeley WW, Butovsky O, Barres BA, Paul SM, Holtzman DM. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017 Sep 28;549(7673):523-527. doi: 10.1038/nature24016. Epub 2017 Sep 20. PMID: 28959956; PMCID: PMC5641217.
Antibodies targeting amyloid-β and ApoE
Active and passive immunization against the Aβ peptide remains a potential future therapy to most likely delay the onset/prevent and possibly treat very mild dementia in AD. My laboratory has been involved in utilizing animal models of Aβ deposition to demonstrate that passive administration with certain antibodies to Aβ and to apoE have potential as a therapy against AD, characterizing the mechanism of action of different anti-Aβ and anti-apoE antibodies, and demonstrating the potential utilization of antibodies for diagnostic purposes in AD. More recently, we have found that a non-lipidated form of apoE is present in amyloid plaques and CAA and that an antibody to this form of apoE removes amyloid plaques similarly to several anti-Aβ antibodies and decreases CAA and improves vascular function to a greater extent. All the work referenced below was carried out in my lab and all experiments, personnel, data analysis, and writing of manuscripts were under my direction.
Publications

APOE Antibody Inhibits Aβ-Associated Tau Seeding and Spreading in a Mouse Model
June 1, 2022
Gratuze M, Jiang H, Wang C, Xiong M, Bao X, Holtzman DM. APOE Antibody Inhibits Aβ-Associated Tau Seeding and Spreading in a Mouse Model. Ann Neurol. 2022 Jun;91(6):847-852. doi: 10.1002/ana.26351. Epub 2022 Mar 31. PMID: 35285073; PMCID: PMC9285984.

APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function
February 17, 2021
Xiong M, Jiang H, Serrano JR, Gonzales ER, Wang C, Gratuze M, Hoyle R, Bien-Ly N, Silverman AP, Sullivan PM, Watts RJ, Ulrich JD, Zipfel GJ, Holtzman DM. APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci Transl Med. 2021 Feb 17;13(581):eabd7522. doi: 10.1126/scitranslmed.abd7522. PMID: 33597265; PMCID: PMC8128342.

Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation
May 1, 2018
Liao F, Li A, Xiong M, Bien-Ly N, Jiang H, Zhang Y, Finn MB, Hoyle R, Keyser J, Lefton KB, Robinson GO, Serrano JR, Silverman AP, Guo JL, Getz J, Henne K, Leyns CE, Gallardo G, Ulrich JD, Sullivan PM, Lerner EP, Hudry E, Sweeney ZK, Dennis MS, Hyman BT, Watts RJ, Holtzman DM. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J Clin Invest. 2018 May 1;128(5):2144-2155. doi: 10.1172/JCI96429. Epub 2018 Mar 30. PMID: 29600961; PMCID: PMC5919821.

Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer’s disease
March 2, 2002
DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science. 2002 Mar 22;295(5563):2264-7. doi: 10.1126/science.1067568. PMID: 11910111.
Synaptic activity and amyloid-β
We found levels of soluble, monomeric Aβ peptide in the brain in vivo, in both mice and humans, are directly regulated by synaptic activity, specifically, by synaptic vesicle release. This finding, with our papers below, strongly suggests that the reason Aβ deposition in the human brain occurs first and to the greatest extent in regions of the brain overlapping with what is called the “default mode network” is due to greater metabolic and synaptic activity in these regions over a lifetime resulting in higher monomeric Aβ levels leading to earlier Aβ aggregation in these regions. All work referenced below was carried out in my lab.
Publications

Neuronal activity regulates the regional vulnerability to amyloid-β deposition
June 1, 2011
Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci. 2011 Jun;14(6):750-6. doi: 10.1038/nn.2801. Epub 2011 May 1. PMID: 21532579; PMCID: PMC3102784.

Amyloid-beta dynamics correlate with neurological status in the injured human brain
August 29, 2008
Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, Zipfel GJ, Holtzman DM. Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science. 2008 Aug 29;321(5893):1221-4. doi: 10.1126/science.1161591. PMID: 18755980; PMCID: PMC2577829.

Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo
April 10, 2008
Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008 Apr 10;58(1):42-51. doi: 10.1016/j.neuron.2008.02.003. PMID: 18400162; PMCID: PMC2390913.

Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo
December 22, 2005
Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005 Dec 22;48(6):913-22. doi: 10.1016/j.neuron.2005.10.028. PMID: 16364896.
Effects of sleep-wake cycle on changes in Aβ, tau, and microglia
We found that in the brain ISF in mice and in CSF in humans, Aβ is higher during wakefulness and lower during sleep. We also found that at least part of these fluctuations are due to differences in synaptic activity and Aβ generation between the wake and sleep states. Increasing wakefulness by sleep deprivation and with orexin both acutely increased Aβ as well as chronically increased Aβ deposition. Increasing sleep with an orexin receptor antagonist acutely decreased Aβ and chronically decreased Aβ deposition. We also found that genetic manipulation of orexin produces similar results and that this effect appears to be due to the effect of orexin on sleep. Once Aβ deposition occurs, this results in disrupted sleep and even greater Aβ production. We also found that wakefulness and sleep deprivation regulates the release of extracellular tau and tau spreading in both mice and humans and that tau pathology is linked with decreased NREM slow wave sleep in humans. Recent work also demonstrates that sleep disruption also affects Aβ deposition and Aβ-induced tau seeding and spreading via the innate immune response/microglia. This work has important implications for not only a mechanism of how normal brain function and its dysfunction could predispose to AD but also suggests that understanding and treating sleep disorders may provide a new way to decrease AD risk. All work below was carried out in my lab.
Publications

APOE-ε4 synergizes with sleep disruption to accelerate Aβ deposition and Aβ-associated tau seeding and spreading
July 17, 2023
Wang C, Nambiar A, Strickland MR, Lee C, Parhizkar S, Moore AC, Musiek ES, Ulrich JD, Holtzman DM. APOE-ε4 synergizes with sleep disruption to accelerate Aβ deposition and Aβ-associated tau seeding and spreading. J Clin Invest. 2023 Jul 17;133(14):e169131. doi: 10.1172/JCI169131. PMID: 37279069; PMCID: PMC10351966.

Sleep deprivation exacerbates microglial reactivity and Aβ deposition in a TREM2-dependent manner in mice
April 26, 2023
Parhizkar S, Gent G, Chen Y, Rensing N, Gratuze M, Strout G, Sviben S, Tycksen E, Zhang Q, Gilmore PE, Sprung R, Malone J, Chen W, Remolina Serrano J, Bao X, Lee C, Wang C, Landsness E, Fitzpatrick J, Wong M, Townsend R, Colonna M, Schmidt RE, Holtzman DM. Sleep deprivation exacerbates microglial reactivity and Aβ deposition in a TREM2-dependent manner in mice. Sci Transl Med. 2023 Apr 26;15(693):eade6285. doi: 10.1126/scitranslmed.ade6285. Epub 2023 Apr 26. PMID: 37099634; PMCID: PMC10449561.
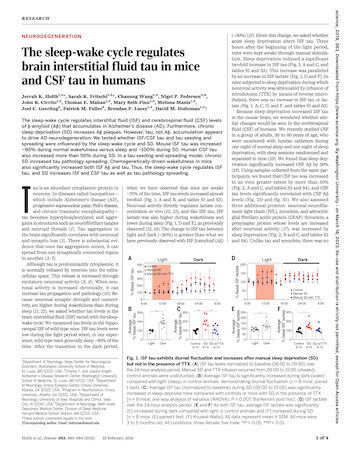
The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans
February 22, 2019
Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, Finn MB, Manis M, Geerling JC, Fuller PM, Lucey BP, Holtzman DM. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science. 2019 Feb 22;363(6429):880-884. doi: 10.1126/science.aav2546. Epub 2019 Jan 24. PMID: 30679382; PMCID: PMC6410369.

Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle
November 13, 2009
Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009 Nov 13;326(5955):1005-7. doi: 10.1126/science.1180962. Epub 2009 Sep 24. PMID: 19779148; PMCID: PMC2789838.
Effects of microglia on Aβ-mediated tau seeding and spreading and tau-mediated neurodegeneration
Increasing evidence suggests that tau pathology spreads from initially affected regions to synaptically connected regions. Once tau aggregates and accumulates, this is directly linked with loss of synapses and neurons (neurodegeneration). We have been able to characterize extracellular tau metabolism utilizing in vivo microdialysis to determine what influences interstitial fluid tau levels. We found that excitatory synaptic activity increases extracellular tau in vivo. In recent work, we have found that microglia are key effectors of both Aβ-induced tau spreading in the brain as well as tau-mediated neurodegeneration. Key molecules and processes that influence these effects of microglia are TREM2, apoE, and the microbiome. All work referenced below were carried out in my lab.
Publications

APOE3ch alters microglial response and suppresses Aβ-induced tau seeding and spread
January 18, 2024
Chen Y, Song S, Parhizkar S, Lord J, Zhu Y, Strickland MR, Wang C, Park J, Tabor GT, Jiang H, Li K, Davis AA, Yuede CM, Colonna M, Ulrich JD, Holtzman DM. APOE3ch alters microglial response and suppresses Aβ-induced tau seeding and spread. Cell. 2024 Jan 18;187(2):428-445.e20. doi: 10.1016/j.cell.2023.11.029. Epub 2023 Dec 11. PMID: 38086389; PMCID: PMC10842861.

Amelioration of Tau and ApoE4-linked glial lipid accumulation and neurodegeneration with an LXR agonist
November 16, 2023
Litvinchuk A, Suh JH, Guo JL, Lin K, Davis SS, Bien-Ly N, Tycksen E, Tabor GT, Remolina Serrano J, Manis M, Bao X, Lee C, Bosch M, Perez EJ, Yuede CM, Cashikar AG, Ulrich JD, Di Paolo G, Holtzman DM. Amelioration of Tau and ApoE4-linked glial lipid accumulation and neurodegeneration with an LXR agonist. Neuron. 2023 Nov 16:S0896-6273(23)00804-8. doi: 10.1016/j.neuron.2023.10.023. Epub ahead of print. PMID: 37995685.

TREM2-independent microgliosis promotes tau-mediated neurodegeneration in the presence of ApoE4
January 18, 2023
Gratuze M, Schlachetzki JCM, D’Oliveira Albanus R, Jain N, Novotny B, Brase L, Rodriguez L, Mansel C, Kipnis M, O’Brien S, Pasillas MP, Lee C, Manis M, Colonna M, Harari O, Glass CK, Ulrich JD, Holtzman DM. TREM2-independent microgliosis promotes tau-mediated neurodegeneration in the presence of ApoE4. Neuron. 2023 Jan 18;111(2):202-219.e7. doi: 10.1016/j.neuron.2022.10.022. Epub 2022 Nov 10. PMID: 36368315; PMCID: PMC9852006.

ApoE isoform- and microbiota-dependent progression of neurodegeneration in a mouse model of tauopathy
January 13, 2023
Seo DO, O’Donnell D, Jain N, Ulrich JD, Herz J, Li Y, Lemieux M, Cheng J, Hu H, Serrano JR, Bao X, Franke E, Karlsson M, Meier M, Deng S, Desai C, Dodiya H, Lelwala-Guruge J, Handley SA, Kipnis J, Sisodia SS, Gordon JI, Holtzman DM. ApoE isoform- and microbiota-dependent progression of neurodegeneration in a mouse model of tauopathy. Science. 2023 Jan 13;379(6628):eadd1236. doi: 10.1126/science.add1236. Epub 2023 Jan 13. PMID: 36634180; PMCID: PMC9901565.

Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model
November 1, 2019
Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, Hoyle R, Holtzman DM. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med. 2019 Nov 4;216(11):2546-2561. doi: 10.1084/jem.20190980. Epub 2019 Oct 10. PMID: 31601677; PMCID: PMC6829593.

TREM2 function impedes tau seeding in neuritic plaques
August 1, 2019
Leyns CEG, Gratuze M, Narasimhan S, Jain N, Koscal LJ, Jiang H, Manis M, Colonna M, Lee VMY, Ulrich JD, Holtzman DM. TREM2 function impedes tau seeding in neuritic plaques. Nat Neurosci. 2019 Aug;22(8):1217-1222. doi: 10.1038/s41593-019-0433-0. Epub 2019 Jun 24. PMID: 31235932; PMCID: PMC6660358.